 Phenylketonurie (PKU)
ist eine komplexe angeborene Stoffwechselstörung, die durch eine genetische Veranlagung verursacht wird, da sie
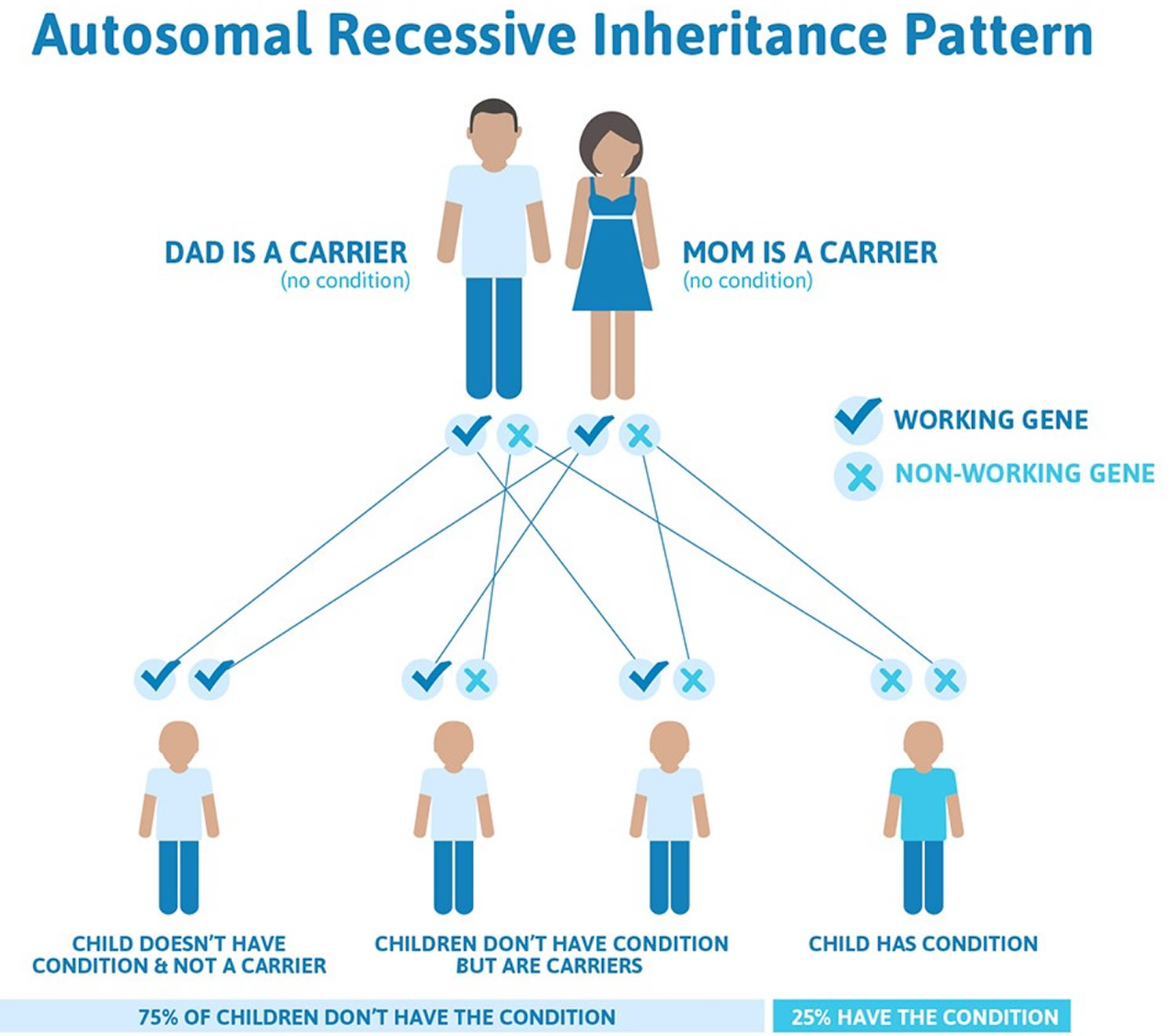
eine autosomal rezessive Vererbung aufweist. Diese Erkrankung wurde erstmals im Jahr 1934 von einem norwegischen
Biochemiker beschrieben und hat seitdem das Interesse von Forschern und Medizinern weltweit auf sich gezogen.
Phenylketonurie (PKU)
ist eine komplexe angeborene Stoffwechselstörung, die durch eine genetische Veranlagung verursacht wird, da sie
eine autosomal rezessive Vererbung aufweist. Diese Erkrankung wurde erstmals im Jahr 1934 von einem norwegischen
Biochemiker beschrieben und hat seitdem das Interesse von Forschern und Medizinern weltweit auf sich gezogen.
Bei PKU ist der Körper nicht in der Lage, Phenylalanin ordnungsgemäß abzubauen, was zu einem Anstieg dieses Stoffes im Blut führt. Dieser Überschuss kann schwerwiegende Auswirkungen auf das zentrale Nervensystem haben. Um die Auswirkungen von PKU zu minimieren, ist eine strenge Diät erforderlich, die phenylalaninarme Lebensmittel einschließt. Zusätzlich ist eine regelmäßige Überwachung des Phenylalaninspiegels im Blut unerlässlich, um sicherzustellen, dass er innerhalb akzeptabler Grenzen bleibt.
Eine frühzeitige Diagnose von PKU ist entscheidend und kann durch Neugeborenenscreening erreicht werden, das routinemäßig in vielen Ländern durchgeführt wird. Dies ermöglicht eine sofortige Intervention, um die Auswirkungen der Krankheit zu mildern und mögliche Komplikationen zu verhindern.
Ohne eine rechtzeitige Intervention kann PKU schwerwiegende Auswirkungen haben, darunter geistige Entwicklungsverzögerung, Hautausschlag oder Hautveränderungen, Mikrozephalie (ein kleiner Kopfumfang), unkontrollierbare Bewegungen und Entwicklungstörungen. Die Symptome können variieren und sich im Laufe der Zeit verschlimmern, was eine lebenslange Betreuung und Überwachung erfordert.
Obwohl es keine Heilung für PKU gibt, kann eine rechtzeitige Diagnose und eine strikte Diät dazu beitragen, die Symptome zu kontrollieren und die Lebensqualität der Betroffenen zu verbessern. Fortschritte in der medizinischen Forschung könnten in Zukunft weitere Behandlungsmöglichkeiten bieten, aber bis dahin bleibt die präventive Überwachung und Betreuung von entscheidender Bedeutung für die Bewältigung dieser komplexen Erkrankung